In this tutorial, we will learn the basic protein data structure used in TorchProtein. In TorchProtein, a protein can be seen as a special case of the general graph in TorchDrug, since either the primary structure (i.e., amino acid sequence) or the tertiary structure (i.e., 3D folded structure) of a protein can be viewed as a graph with atom or residue as nodes and different edge construction methods.
Before we start, you are suggested to first read the Notes on Graph Data Structures in TorchDrug.
Protein Data Structure I/O
Typically, we can get the protein structure information from a PDB file,
which is a standard data format that describes the protein structure.
In this tutorial, we use the single-chain Insulin (PDB id: 2LWZ) as an example.
Let’s first visualize it via NGLView.
import nglview
view = nglview.show_pdbid("2lwz")
view
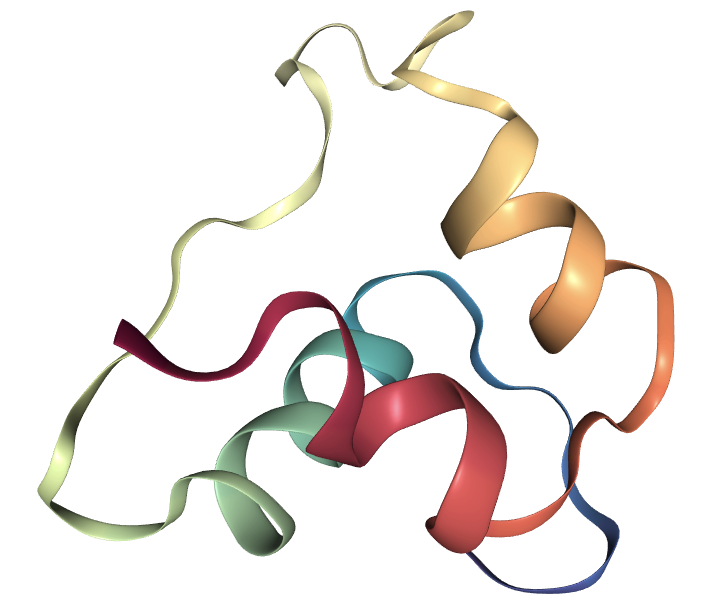
Construct Protein Data Structure from PDB File
In TorchProtein, we can use Protein.from_pdb to read the PDB file and construct the data structure.
The atom, edge and residue features may serve as input to machine learning models.
We can specify different features by changing the arguments in Protein.from_pdb.
import torchdrug as td
from torchdrug import data, utils
pdb_file = utils.download("https://files.rcsb.org/download/2LWZ.pdb", "./")
protein = data.Protein.from_pdb(pdb_file, atom_feature="position", bond_feature="length", residue_feature="symbol")
print(protein)
print(protein.residue_feature.shape)
print(protein.atom_feature.shape)
print(protein.bond_feature.shape)
Protein(num_atom=445, num_bond=916, num_residue=57)
torch.Size([57, 21])
torch.Size([445, 3])
torch.Size([916, 1])
The constructed data structure contains rich information about the protein. For example, you can get the chain ids of the first 10 residues and the 3D coordinates of the first 10 atoms as below.
for residue_id, chain_id in zip(protein.residue_type.tolist()[:10], protein.chain_id.tolist()[:10]):
print("%s: %s" % (data.Protein.id2residue[residue_id], chain_id))
for atom, position in zip(protein.atom_name.tolist()[:10], protein.node_position.tolist()[:10]):
print("%s: %s" % (data.Protein.id2atom_name[atom], position))
The protein data structure stores all information needed to recover a protein and provides a to_pdb() method to save the protein in PDB format.
We show the recovery of the single-chain Insulin as below.
from rdkit import Chem
protein.to_pdb("new_2LWZ.pdb")
mol = Chem.MolFromPDBFile("new_2LWZ.pdb")
view = nglview.show_rdkit(mol)
view
Construct Protein Data Structure from Protein Sequence
In some applications, we may only access the amino acid sequence of the protein.
For such cases, TorchProtein provide a Protein.from_sequence method and a Protein.from_sequence_fast method to construct the protein data structure from sequence.
The former method constructs the protein object using RDKit, which will compute atom, residue and bond features and is thus slower.
The latter method directly constructs the protein data structure with only residue types and features and is thus much faster.
import time
aa_seq = protein.to_sequence()
print(aa_seq)
start_time = time.time()
seq_protein = data.Protein.from_sequence(aa_seq, atom_feature="symbol", bond_feature="length", residue_feature="symbol")
end_time = time.time()
print("Duration of construction: ", end_time - start_time)
print(seq_protein)
start_time = time.time()
seq_protein = data.Protein.from_sequence(aa_seq, atom_feature=None, bond_feature=None, residue_feature="default")
end_time = time.time()
print("Duration of construction: ", end_time - start_time)
print(seq_protein)
FVNQHLCGSDLVEALYLVCGERGFFYTDPTGGGPRRGIVEQCCHSICSLYQLENYCN
Duration of construction: 0.5593459606170654
Protein(num_atom=445, num_bond=910, num_residue=57)
Duration of construction: 0.0017361640930175781
Protein(num_atom=0, num_bond=0, num_residue=57)
Protein Operations
Batch Protein
To fully utilize the hardware, TorchProtein inherits from the data.Graph structure in TorchDrug and supports to process multiple proteins as a batch, and the batch can switch between CPUs and GPUs using cpu() and cuda() methods.
Given multiple proteins, we can construct the protein batch via data.Protein.pack and transfer it from CPU to GPU via cuda().
Also, we can extract several specific proteins from the batch by the normal indexing operation.
proteins = [protein] * 3
proteins = data.Protein.pack(proteins)
print(proteins)
proteins = proteins.cuda()
print(proteins)
proteins_ = proteins[[0, 2]]
print(proteins_)
PackedProtein(batch_size=3, num_atoms=[445, 445, 445], num_bonds=[916, 916, 916], num_residues=[57, 57, 57])
PackedProtein(batch_size=3, num_atoms=[445, 445, 445], num_bonds=[916, 916, 916], num_residues=[57, 57, 57], device='cuda:0')
PackedProtein(batch_size=2, num_atoms=[445, 445], num_bonds=[916, 916], num_residues=[57, 57], device='cuda:0')
References between Atoms and Residues
In TorchProtein, we provide the atom2residue method to retrieve the corresponding residue of each atom,
and provide the residue2atom method to retrieve the associated atoms of each residue.
Typical usages of these two methods are as below.
for atom_id, (atom, residue_id) in enumerate(zip(protein.atom_name.tolist()[:20], protein.atom2residue.tolist()[:20])):
print("[atom %s] %s: %s" % (atom_id, data.Protein.id2atom_name[atom], data.Protein.id2residue[residue_id]))
for residue_id in [0, 1]:
atom_ids = protein.residue2atom(residue_id).sort()[0]
for atom, position in zip(protein.atom_name[atom_ids].tolist(), protein.node_position[atom_ids].tolist()):
print("[residue %s] %s: %s" % (residue_id, data.Protein.id2atom_name[atom], position))
Subprotein and Masking
In protein research, we sometimes need to extract specific residues from a protein and analyze them. With TorchProtein, we can easily achieve this by the indexing operation. We give an example of extracting the first two residues from a protein as below. Note that, during the extraction, the bonds between the atoms of the extracted residues will preserve.
first_two = protein[:2]
first_two.visualize()
In TorchProtein, we also provide the resiude_mask method to extract some specified residues from a protein
and provide the node_mask method to extract some specified atoms from a protein.
By using these two methods, we can also extract the first two residues from a protein as below.
is_first_two_ = (protein.residue_number == 1) | (protein.residue_number == 2)
first_two_ = protein.residue_mask(is_first_two_, compact=True)
assert first_two == first_two_
is_first_two_ = (protein.atom2residue == 0) | (protein.atom2residue == 1)
first_two_ = protein.node_mask(is_first_two_, compact=True)
assert first_two == first_two_
Atom and Residue Views
For sequence-based protein encoding models, we typically see residues as nodes in a protein graph
while sometimes we also want to use atom features as node features for structure-based protein encoding models.
To support flexible switch between atom and residue features, TorchProtein defines the view attribute to select which features we want to use as node features.
protein.view = "atom"
print(protein.node_feature.shape)
protein.view = "residue"
print(protein.node_feature.shape)
torch.Size([445, 3])
torch.Size([57, 21])
Register Your Own Attributes
While the Protein class comes with several atom- and residue-level attributes, we may also want to define our own attributes. This only requires to wrap the attribute assignment lines with a context manager.
We can use protein.atom(), protein.residue() and protein.graph() for atom-, residue- and graph-level attributes, respectively.
Register Residue and Atom Attributes
We give two examples of registering residue and atom attributes here. The first example defines a custom residue attribute to encode whether each residue is followed by a residue of “GLY”. The second example defines a custom atom attribute to encode whether each atom is connected to a nitrogen.
from torch_scatter import scatter_add
next_residue_type = torch.cat([protein.residue_type[1:], torch.full((1,), -1, dtype=protein.residue_type.dtype)])
followed_by_GLY = next_residue_type == data.Protein.residue2id["GLY"]
with protein.residue():
protein.followed_by_GLY = followed_by_GLY
atom_in, atom_out = protein.edge_list.t()[:2]
attached_to_N = scatter_add(protein.atom_type[atom_in] == td.NITROGEN, atom_out, dim_size=protein.num_node)
with protein.atom():
protein.attached_to_N = attached_to_N
Register References between Residues and Atoms
In some cases, we would like to link a residue/atom to another residue/atom.
We can achieve this by registering under the context of protein.residue_reference() or protein.atom_reference().
For example, we can register the index of the corresponding alpha carbon of each residue under the context of protein.residue() and protein.atom_reference().
Note that, under any operation of extracting a part of a protein, the indices registered in this way will automatically change to the indices under the new extracted protein.
from torch_scatter import scatter_max
range = torch.arange(protein.num_node)
calpha = torch.where(protein.atom_name == protein.atom_name2id["CA"], range, -1)
residue2calpha = scatter_max(calpha, protein.atom2residue, dim_size=protein.num_residue)[0]
with protein.residue(), protein.atom_reference():
protein.residue2calpha = residue2calpha
sub_protein = protein[3:10]
for calpha_index in sub_protein.residue2calpha.tolist():
atom_name = data.Protein.id2atom_name[sub_protein.atom_name[calpha_index].item()]
print("New index %d: %s" % (calpha_index, atom_name))
New index 1: CA
New index 10: CA
New index 20: CA
New index 28: CA
New index 34: CA
New index 38: CA
New index 44: CA