Unified Protein Data Structures
To broadly facilitate the usage of protein sequences, protein structures and both of them, TorchProtein designs a unified data structure to represent protein data. It allows you to seamlessly switch between the sequence and structure views of a protein object and apply machine learning models and graph construction methods upon it.
pdb_file = utils.download("https://files.rcsb.org/download/2LWZ.pdb", "./")
protein = data.Protein.from_pdb(pdb_file, atom_feature="position", bond_feature="length", residue_feature="symbol")
graph_construction_model = layers.GraphConstruction(node_layers=[geometry.AlphaCarbonNode()],
edge_layers=[geometry.SpatialEdge(distance=10.0, sequence_distance=5)])
model = models.GearNet(input_dim=21, hidden_dims=[512, 512, 512], num_relation=1, readout="sum")
processed_protein = graph_construction_model(data.Protein.pack([protein]))
output = model(processed_protein, processed_protein.residue_feature.float())
aa_seq = protein.to_sequence()
seq_protein = data.Protein.from_sequence(aa_seq, atom_feature="symbol", bond_feature="length", residue_feature="symbol")
seq_model = models.ProteinCNN(input_dim=21, hidden_dims=[1024, 1024], kernel_size=5, padding=2, readout="max")
seq_output = seq_model(data.Protein.pack([seq_protein]), data.Protein.pack([seq_protein]).residue_feature.float())
Flexible Building Blocks
Equipped with a large collection of datasets and building blocks, it is easy to instantiate dataset and build standard models in TorchProtein without writing boilerplate code. These building blocks are highly flexible and extensible to boost exploration of model designs.
transform = transforms.ProteinView(view="residue")
ec = datasets.EnzymeCommission("~/protein-datasets/", transform=transform)
graph_construction_model = layers.GraphConstruction(node_layer=geometry.AlphaCarbonNode(),
edge_layers=[geometry.SpatialEdge(distance=10.0, sequence_distance=5)])
model = layers.Sequential(
layers.GeometricRelationalGraphConv(ec.residue_feature_dim, 512, num_relation=1),
layers.GeometricRelationalGraphConv(512, 512, num_relation=1),
layers.SumReadout(),
global_args=("graph",)
)
protein = ec[0]["graph"]
processed_protein = graph_construction_model(data.Protein.pack([protein]))
feature = model(processed_protein, processed_protein.node_feature.float())
Extensive Benchmarks
Based on TorchProtein, we build the comprehensive PEER benchmark which contains 14 protein sequence understanding tasks lying in 5 task groups. On this benchmark, we systematically compare between different protein sequence encoding methods. The benchmark will be continually extended to incorporate important benchmark tasks and track new sequence-based models. The benchmark leaderboards will be maintained to manifest the relative strength between different methods. Here, we visualize the benchmark results of single-task learning methods on three types of tasks.
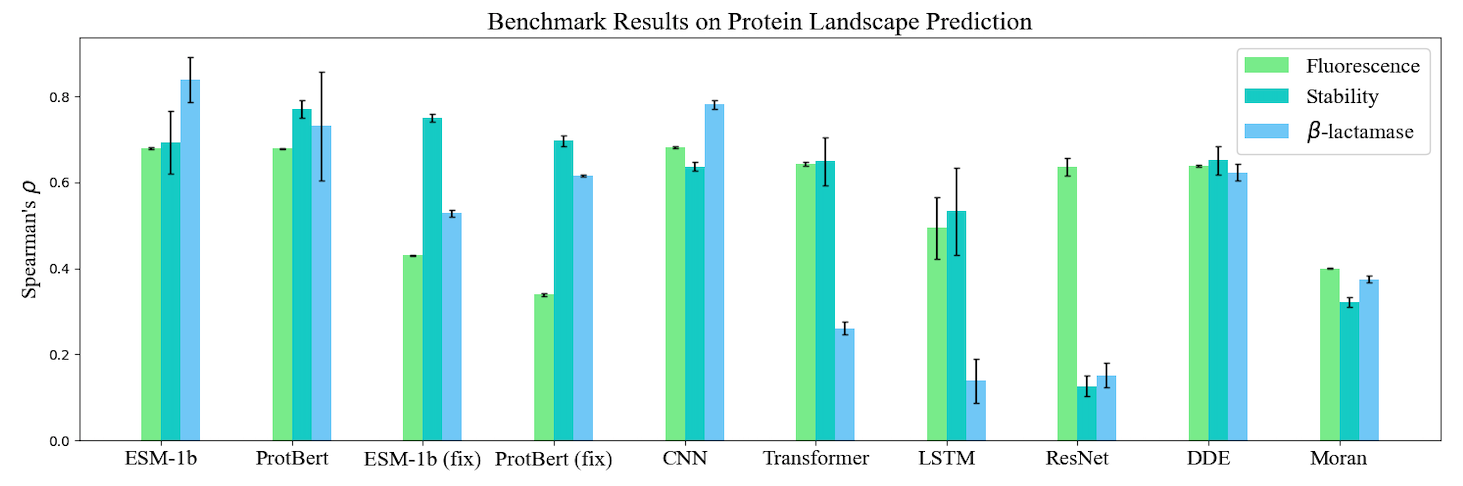
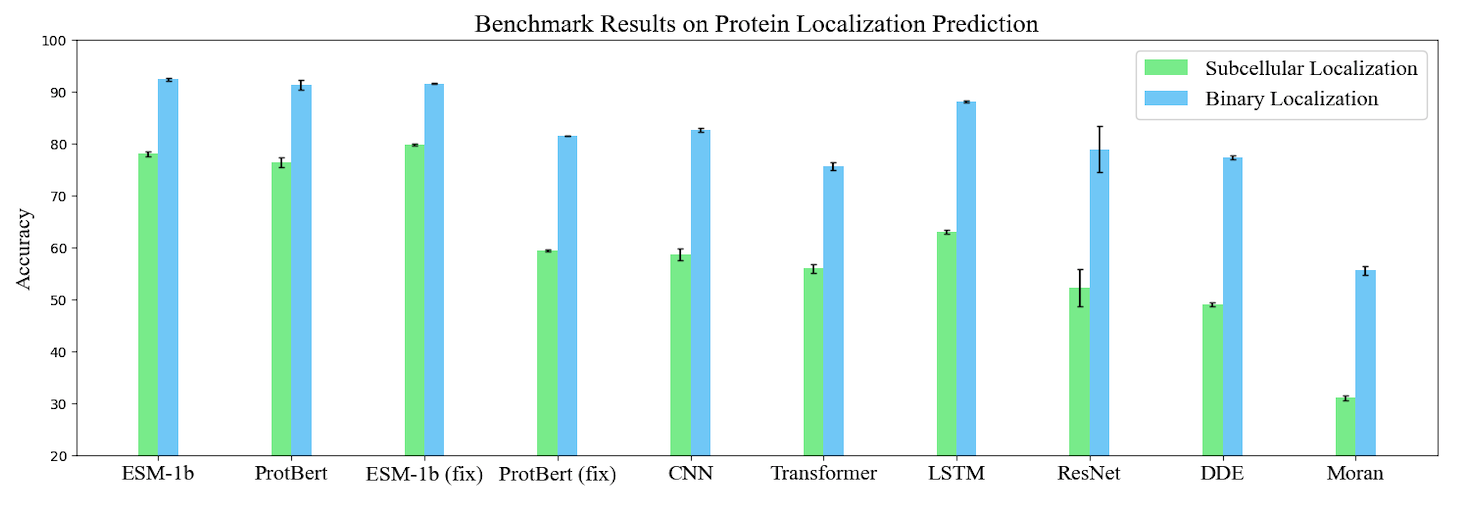
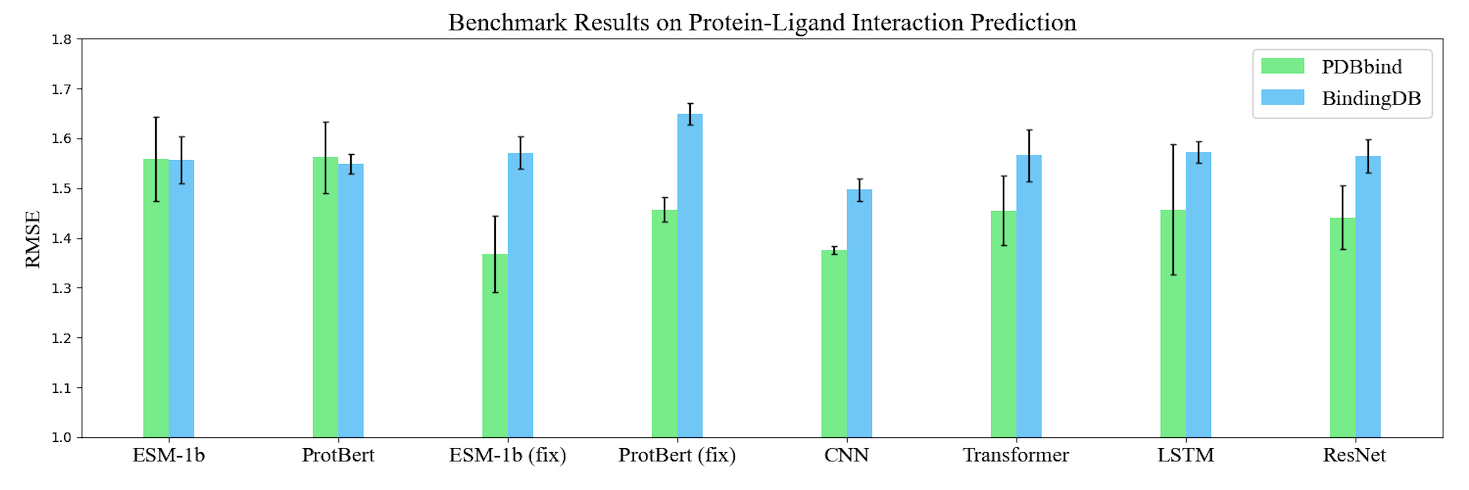
Protein Model Zoo
Coming soon!